

コハク酸デスベンラファキシンは、小児集団への使用を適応としていません。

デスベンラファキシンコハク酸塩一水和物の禁忌 – メドレー
デスベンラファクシンコハク酸塩一水和物、ベンラファクシン塩酸塩、または製剤中の任意の賦形剤に対する過敏症。
デスベンラファクシンは、セロトニンおよびノルエピネフリン再取り込み阻害剤です。コハク酸デスベンラファキシンは、モノアミンオキシダーゼ阻害剤 (MAOI) と併用したり、MAOI による治療を中止してから少なくとも 14 日後には使用しないでください。コハク酸デスベンラファクシン一水和物の半減期に基づくと、コハク酸デスベンラファクシンの中止後、MAOI を開始する前に少なくとも 7 日間待つ必要があります。リネゾリドなどの可逆的MAOIで治療されている患者、または静脈内メチレンブルーが投与されている患者にコハク酸デスベンラファクシンの投与を開始することも、セロトニン症候群のリスクが増加するため禁忌です。
この薬は小児への使用は禁忌です。
デスベンラファキシンコハク酸塩一水和物の使用方法 – メドレー
この薬は割ったり、開けたり、噛んだりしないでください。
コハク酸デスベンラファクシンの推奨用量は、食事の有無にかかわらず、1 日 1 回 50 mg です。臨床研究では、50~400 mg/日の用量が効果的であることが示されましたが、50 mg/日を超える用量では他の利点は実証されませんでした。臨床判断に基づいて、一部の患者に用量の増加が必要な場合は、少なくとも 7 日の間隔をあけて徐々に増量する必要があります。最大用量は 200 mg/日を超えてはなりません。
腎不全患者への使用
重度の腎不全(24時間CrCl lt; 30 mL/分)または末期腎疾患(ESRD)の患者における推奨初回用量は、1日おきに50 mgです。これらの患者のクリアランスには個人差があるため、用量の個別化が望ましい場合があります。透析後の患者に追加用量を投与すべきではありません。
肝不全患者への使用
肝障害のある患者には用量調整は必要ありません。ただし、100 mg/日を超える用量増加は推奨されません。
小児への使用
18 歳未満の患者に対する安全性と有効性はまだ確立されていません。
高齢患者への使用
年齢のみに基づいて用量を調整する必要はありません。ただし、使用量を決定する際には、デスベンラファクシンの腎クリアランスの低下の可能性を考慮する必要があります。
一部の高齢患者のデスベンラファクシンに対する感受性の高さは無視できません。
デスベンラファクシンの中止
デスベンラファクシンの中止に伴う症状は、他のSNRIおよびSSRIと同様に報告されています。患者が治療を中止するときは、これらの症状がないか監視する必要があります。可能な限り、突然の中止ではなく、徐々に用量を減らすことが推奨されます。用量の減量または治療の中止後に耐えられない症状が発生した場合は、以前に処方された用量を再開することを検討する必要があります。その後、医師は用量を徐々に減らし続ける場合があります。
他の抗うつ薬による治療をデスベンラファキシンによる治療に置き換える
患者がベンラファクシンを含む他の抗うつ薬からデスベンラファクシンに置き換えられた場合、中止症状が報告されています。中止症状を最小限に抑えるために、最初の抗うつ薬を徐々に中止することが必要な場合があります。
デスベンラファクシンとリネゾリドやメチレンブルーなどの可逆的 MAOI の併用
リネゾリドなどの可逆的MAOIで治療中の患者、またはセロトニン症候群のリスクが高いため、メチレンブルーの静脈内投与を受けている患者にはデスベンラファクシンの投与を開始しないでください。精神疾患のより緊急の治療が必要な患者の場合は、入院を含む非薬理学的介入を考慮する必要があります。
場合によっては、すでにデスベンラファキシン療法を受けている患者には、リネゾリドまたは静脈内メチレンブルーによる緊急治療が必要になる場合があります。リネゾリドまたは静脈内メチレンブルー治療に代わる許容可能な代替手段がなく、特定の患者におけるリネゾリドまたは静脈内メチレンブルー治療の潜在的な利点がセロトニン症候群のリスクを上回る場合、デスベンラファクシンは直ちに中止されるべきであり、デスベンラファクシンも直ちに中止されるべきです。メチレンブルーの静脈内投与が可能です。患者は、2週間、またはリネゾリドまたはメチレンブルーの静脈内投与の最後の投与後24時間のいずれか早い方まで、セロトニン症候群の症状がないか監視される必要があります。デスベンラファクシン療法は、リネゾリドまたは静脈内メチレンブルーの最後の投与から 24 時間後に再開できます。
デスベンラファキシンコハク酸塩一水和物に関する注意事項 – Medley
うつ病の症状の臨床的悪化、行動の異常な変化、および自殺傾向

コハク酸デスベンラファクシンは、うつ病の治療に使用できる薬剤の一種であるSNRI(セロトニンおよびノルエピネフリン再取り込み阻害剤)です。デスベンラファクシンで治療されたすべての患者は、臨床症状の悪化や自殺傾向について適切に監視され、注意深く観察されるべきです。患者、その家族、およびその介護者は、不安、動揺、パニック発作、不眠症、イライラ、敵意、攻撃性、衝動性、アカシジア(精神運動性興奮)、軽躁状態、躁状態、その他の異常な行動の変化の出現に注意するよう奨励されるべきである。うつ病や自殺念慮の悪化、特に治療開始時や用量や投与スケジュールの変更時。特にうつ病患者では、自殺未遂のリスクを考慮する必要があり、過剰摂取のリスクを減らすために、適切な患者ケアと両立できる最小限の薬剤を投与する必要があります。
自殺はうつ病やその他の精神疾患のリスクとして知られており、これらの疾患だけでも自殺の強力な予測因子となります。抗うつ薬(SSRI(選択的セロトニン再取り込み阻害剤)など)の短期プラセボ対照研究の統合分析により、これらの薬剤が小児、青少年、若年成人(18~24歳)の自殺リスクを高めることが実証された。 )大うつ病およびその他の精神疾患を患っている。短期研究では、24 歳以上の成人において、プラセボと比較して抗うつ薬による自殺リスクの増加は証明されていません。 65歳以上の成人では、抗うつ薬を服用した場合、プラセボを服用した場合と比較して自殺のリスクが減少しました。
躁状態/軽躁状態
臨床研究では、デスベンラファクシンで治療された患者の0.03%で躁病が報告されました。他の市販の抗うつ薬で治療を受けている重度の情動障害患者の少数において、躁状態/軽躁状態の活性化も報告されています。すべての抗うつ薬と同様に、デスベンラファクシンは躁病または軽躁病の個人歴や家族歴のある患者には注意して使用する必要があります。
セロトニン症候群または神経弛緩薬悪性症候群 (NMS) に類似した反応
他のセロトニン作動薬と同様、デスベンラファクシンによる治療、特に他のセロトニン作動薬(SSRI、SNRI、トリプタンを含む)との併用では、致死性の可能性があるセロトニン症候群または神経弛緩性悪性症候群(NMS)様反応が発生する可能性があります。セロトニン代謝を損なう薬物療法(例、リネゾリドや静脈内メチレンブルーなどの可逆的MAOIを含むMAOI)、または抗精神病薬や他のドーパミン拮抗薬との併用。セロトニン症候群の症状には、精神状態の変化(例、興奮、幻覚、昏睡)、自律神経の不安定(例、頻脈、不安定な血圧、高体温)、神経筋の異常(例:反射亢進、協調運動障害)、および/または胃腸疾患が含まれる場合があります。症状(吐き気、嘔吐、下痢など)。最も重度のセロトニン症候群は、高熱、筋肉の硬直、バイタルサインの急速な変動や精神状態の変化を伴う自律神経の不安定性を含む NMS に似ている場合があります。
デスベンラファクシンと、セロトニン作動性および/またはドーパミン作動性神経伝達物質系に影響を与える可能性のある他の薬剤との併用治療が臨床的に正当である場合、特に治療開始時および用量増加中は患者を注意深く観察することが推奨されます。
デスベンラファクシンとセロトニン前駆体 (トリプトファン サプリメントなど) の併用は推奨されません。
閉塞隅角緑内障
散瞳症はデスベンラファクシンとの関連で報告されています。したがって、眼圧が上昇している患者、または狭隅角/閉塞性緑内障のリスクがある患者は監視する必要があります。
ベンラファクシンおよび/またはデスベンラファクシンを含む薬剤の併用投与
デスベンラファクシンはベンラファクシンの主な活性代謝物です。デスベンラファクシンコハク酸塩一水和物を含む製品は、ベンラファクシン塩酸塩を含む製品、またはデスベンラファクシンコハク酸一水和物を含む他の製品と同時に使用しないでください。
血圧への影響
臨床研究では一部の患者、特に高用量で血圧の上昇が観察されています。デスベンラファキシンによる治療前に、既存の高血圧を管理する必要があります。デスベンラファクシンを投与されている患者は、定期的に血圧をモニタリングする必要があります。即時治療が必要な高血圧の症例がデスベンラファクシンで報告されています。血圧の上昇が続くと悪影響が生じる可能性があります。デスベンラファクシンの投与中に血圧の持続的な上昇を経験した患者の場合は、用量の減量または中止を検討する必要があります。血圧上昇の影響を受ける可能性のある臨床症状のある患者には注意が必要です。
心血管・脳血管
心血管障害、脳血管障害、または脂質代謝障害のある患者にデスベンラファクシンを投与する場合は注意が必要です。デスベンラファクシンを用いた臨床研究では、血圧と心拍数の上昇が観察されました。デスベンラファクシンは、心筋梗塞、不安定な心疾患、コントロール不良の高血圧、脳血管疾患の最近の病歴を持つ患者に対して体系的に評価されていません。脳血管疾患を除くこれらの診断を受けた患者は臨床研究から除外された。
不安定狭心症
デスベンラファクシンは不安定狭心症患者において体系的に評価されていないため、この集団ではその使用は推奨されません。
血清脂質
デスベンラファクシンの用量に関連した血清総コレステロール、LDL(低密度リポタンパク質)コレステロール、およびトリグリセリドの増加が臨床研究で観察されています。血清脂質の定期的な管理は、デスベンラファキシンによる治療中に実行する必要がありますが、この管理の頻度は医師の裁量に任されています。
発作
デスベンラファクシンを用いた臨床研究では発作の症例が報告されています。デスベンラファクシンは、発作性障害患者において体系的に評価されていません。発作の病歴のある患者は臨床研究から除外された。このような患者にはコハク酸デスベンラファキシンを慎重に処方する必要があります。
デスベンラファクシンコハク酸塩治療の中止の影響
SNRI および SSRI の市販中、これらの薬剤の中止時に、特に突然の場合に、次のような有害事象が発生するという報告が自然発生的に報告されています。これには、気分不快、イライラ、興奮、めまい、感覚障害 (例、電気ショック感覚などの感覚異常) が含まれます。 )、不安、混乱、頭痛、無気力、情緒不安定、不眠症、軽躁状態、耳鳴り、発作。これらの事象は通常自然に制限されますが、重篤な中止症状が報告されています。
コハク酸デスベンラファキシンによる治療を中止した場合、患者の症状を監視する必要があります。可能な限り、突然の中止ではなく、徐々に用量を減らすことが推奨されます。用量の減量または治療の中止後に耐えられない症状が発生した場合は、以前に処方された用量を再開することを検討する必要があります。

異常出血
コハク酸デスベンラファクシンを含む SNRI および SSRI は、出血事象のリスクを高める可能性があります。アセチルサリチル酸、非ステロイド性抗炎症薬(NSAID)、ワルファリン、その他の抗凝固薬の併用がこのリスクに寄与する可能性があります。 SNRI および SSRI に関連する出血は、斑状出血、血腫、鼻出血、点状出血から生命を脅かす出血まで多岐にわたります。患者は、デスベンラファクシンコハク酸塩と NSAID、アセチルサリチル酸、または凝固や出血に影響を与えるその他の物質の併用に伴う出血のリスクについて警告される必要があります。
腎不全
中等度または重度の腎不全または末期腎疾患(ESRD)の患者では、デスベンラファクシンコハク酸塩のクリアランスが減少し、その結果、薬物の排出半減期が延長されました。その結果、コハク酸デスベンラファクシンへの曝露が臨床的に有意に増加する可能性がありました。重度の腎障害または ESRD のある患者には、用量の調整 (1 日おきに 50 mg) が必要です。中等度または重度の腎障害または ESRD のある患者では、用量を増加させるべきではありません。
低ナトリウム血症
SNRI(コハク酸デスベンラファキシン一水和物を含む)およびSSRIを使用した低ナトリウム血症および/または抗利尿ホルモン分泌不全症候群(SIADH)の症例は、通常、高齢患者や利尿薬を服用している患者を含む血液量減少または脱水症状の患者で報告されています。
間質性肺疾患と好酸球性肺炎
ベンラファクシン(コハク酸デスベンラファクシンの親薬)による治療に関連した間質性肺疾患および好酸球性肺炎はほとんど報告されていません。デスベンラファクシンコハク酸塩で治療され、進行性の呼吸困難、咳、または胸部不快感を発症した患者では、これらの副作用の可能性を考慮する必要があります。これらの患者は直ちに医学的評価を受ける必要があり、コハク酸デスベンラファキシンの中止を検討する必要があります。
受胎能力、妊娠、授乳
ヒトの妊娠におけるデスベンラファクシンの安全性は確立されていません。デスベンラファクシンは、期待される利益が起こり得るリスクを上回る場合にのみ妊婦に投与されるべきです。デスベンラファクシンを出産まで、または出産直前に使用する場合は、中止による新生児への影響を考慮する必要があります。
妊娠後期に SNRI または SSRI に曝露された新生児では、呼吸補助、経管栄養、長期入院の必要性などの合併症が報告されています。これらの合併症は出生直後に発生する可能性があります。
コハク酸デスベンラファキシンは、妊娠リスクカテゴリー C に分類される医薬品です。したがって、妊娠中の女性は医師または歯科外科医のアドバイスなしにこの薬を使用しないでください。
デスベンラファクシン (O-デスメチルベンラファクシン) は母乳中に排泄されます。授乳中の乳児がデスベンラファクシンに曝露されると重篤な副作用が起こる可能性があるため、母親にとっての薬剤の重要性を考慮して、授乳を中止するか薬剤を中止するかを決定する必要があります。予想される利益が考えられるリスクを上回る場合にのみ、授乳中の女性にデスベンラファクシンを投与します。
小児および高齢者向けの使用
「薬理学的特性 – 薬物動態特性」、「使用方法」、および「反応」の項目を参照してください。
機械を運転および操作する能力への影響 – 認知能力および運動能力への干渉
健康な個人の行動能力に対するデスベンラファクシンの効果を評価した臨床研究の結果では、精神運動能力、認知能力、または複雑な行動能力に臨床的に重大な障害がないことが明らかになりました。ただし、中枢神経系に作用する薬剤は判断力、推論力、または運動能力を損なう可能性があるため、デスベンラファクシン療法が患者の能力に悪影響を及ぼさないことが十分に確信できるまでは、自動車を含む危険な機械の操作について患者に警告する必要があります。活動。
治療中は、能力や注意力が損なわれる可能性があるため、患者は車の運転や機械の操作を行わないでください。
虐待と依存 — 身体的および精神的依存
デスベンラファクシンは、前臨床または臨床研究において乱用の可能性について体系的に研究されていませんが、臨床研究では薬物探索行動の兆候は観察されていません。
この薬はドーピングを引き起こす可能性があります。
デスベンラファキシンコハク酸塩一水和物の副作用 – メドレー
臨床研究の経験
デスベンラファクシンの安全性は、TDM 臨床研究または市販後の経験において、10 ~ 400 mg/日の範囲のデスベンラファクシンを少なくとも 1 回投与された合計 8,453 人の患者において確立されました。長期安全性は、TDM 研究において少なくとも 6 か月間デスベンラファキシンに曝露された 2,140 人の患者で評価され、そのうち 421 人の患者は 1 年間曝露されました。
System Organ Class (SOC) および CIOMS (国際医療機関評議会) の頻度カテゴリー別の副作用を、各頻度カテゴリーおよび SOC 内で医学的重症度が降順にリスト化しています。
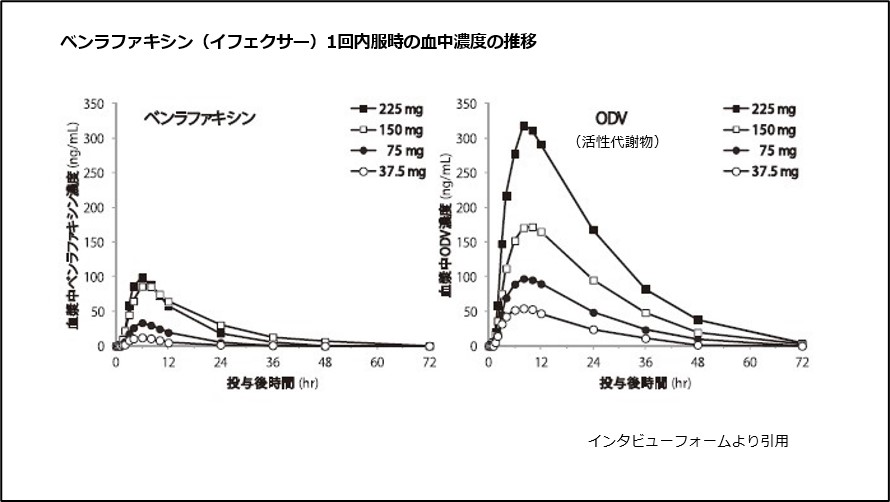
潜在的な抗うつ作用を確認するために、日中に得られた睡眠ポリグラフ(PSG)と脳波(EEG)の記録を使用して健康な人を対象に実施された研究では、デスベンラファクシンは潜時を増加させ、レム睡眠の時間を短縮しました。効果は、1日量150 mg、300 mg、および600 mgの投与で観察されました。
虚血性心臓の有害事象
臨床研究では、心筋虚血、心筋梗塞、血行再建を必要とする冠状動脈閉塞などの虚血性心臓の有害事象がまれに報告されています。これらの患者には複数の潜在的な心臓危険因子があった。プラセボと比較して、デスベンラファクシンによる治療中にこれらの事象を経験した患者の数が多かった。
不安定狭心症
具体的には、閉経後女性の血管運動症状の治療のためのデスベンラファクシンの研究において、200mg/日を超えるデスベンラファクシンの投与を受けた5人の女性で虚血性心臓有害事象が観察された。これらの患者には複数の潜在的な危険因子がありました。
中止の症状
TDM臨床試験における治療の突然の中止、用量の減量、または段階的な中止に関連して報告された副作用は?の割合で報告されています。 2%には、めまい、離脱症候群、吐き気、頭痛が含まれます。一般に、投与量が高く、治療期間が長いほど、中止症状がより頻繁に発生しました。
治療の中止につながる副作用
最長12週間の短期研究でデスベンラファキシンによる治療を受けた患者の少なくとも2%で中止に至った最も一般的な副作用は吐き気(2%)であった。最長 11 か月の長期研究では、少なくとも 2% の患者で中止につながる事象は発生せず、その割合は二重盲検相におけるプラセボよりも高かった。
高齢者向けの使用
デスベンラファキシンによる治療を受けた MDD 臨床研究の患者 7,785 人のうち、患者の 5% は 65 歳以上でした。これらの患者と若い患者の間で、安全性または有効性における全体的な差異は観察されませんでした。しかし、短期のプラセボ対照研究では、収縮期起立性低血圧の発生率が増加し、短期および長期のプラセボ対照研究の両方で、患者の収縮期血圧の上昇が見られました。 LT患者と比較すると65歳。デスベンラファクシンによる治療を受けた65歳。
有害事象が発生した場合は、健康監視通知システム (NOTIVISA) または州または地方自治体の健康監視機関に通知してください。
デスベンラファキシンコハク酸塩一水和物の組成 – メドレー
各徐放性フィルムコーティング錠には次のものが含まれます。
75.840 mgのデスベンラファクシンコハク酸塩一水和物、50 mgのデスベンラファクシン塩基に相当します。
賦形剤:
微結晶セルロース、ヒプロメロース、二酸化ケイ素、タルク、ステアリン酸マグネシウム、精製水、ポリビニルアルコール、二酸化チタン、マクロゴール、ベンガラ。
各徐放性フィルムコーティング錠には次のものが含まれます。
151.67 mgのデスベンラファクシンコハク酸塩一水和物、100 mgのデスベンラファクシン塩基に相当します。
賦形剤:
微結晶セルロース、ヒプロメロース、二酸化ケイ素、タルク、ステアリン酸マグネシウム、精製水、ポリビニルアルコール、二酸化チタン、マクロゴール、ベンガラ、黄色酸化鉄。
デスベンラファキシンコハク酸塩一水和物のプレゼンテーション – メドレー
50mg錠
7、14、28錠。
100mg錠
7、14、28錠。
デスベンラファキシンコハク酸塩一水和物の薬物相互作用 – メドレー
モノアミンオキシダーゼ阻害剤 (MAOI)
副作用の一部は重篤なもので、最近モノアミンオキシダーゼ阻害剤(リネゾリドや静脈内メチレンブルーなどの可逆的MAOIを含む)を中止し、デスベンラファクシンと同様の薬理特性を持つ抗うつ薬(SNRIまたはSSRI)による治療を開始した患者で報告されています。 MAOIを開始する前に最近SNRIまたはSSRI療法を中止した人。モノアミンオキシダーゼ阻害剤(MAOI)を服用している患者におけるデスベンラファクシンの併用は禁忌です。
MAOI の中止とコハク酸デスベンラファキシン療法の導入の間には、少なくとも 14 日間経過する必要があります。さらに、コハク酸デスベンラファクシンの中止後、MAOI を開始する前に少なくとも 7 日間の休憩が必要です。
中枢神経系 (CNS) 活性剤
デスベンラファクシンを他の CNS 活性薬剤と組み合わせて使用するリスクは体系的に評価されていません。デスベンラファキシンを他のCNS活性薬と組み合わせて服用する場合は注意が必要です。
セロトニン作動性症候群
他のセロトニン作動薬と同様に、デスベンラファクシンによる治療、特にセロトニン神経伝達物質系に影響を与える可能性のある他の薬剤(トリプタン、SSRI、他のSNRI、リチウム、シブトラミン、フェンタニル、およびその類似体、トラマドール、デキストロメトルファン、タペンタドール、メペリジン、メサドン、ペンタゾシンまたはセントジョーンズワート[オトギリソウ])、セロトニン代謝を損なう薬剤(リネゾリド[可逆的非選択的MAOIである抗生物質]を含むMAOIなど)と併用およびメチレンブルー)またはセロトニン前駆体(トリプトファンサプリメントなど)を使用します。
デスベンラファクシンと SSRI、SNRI、または 5-ヒドロキシトリプタミン [トリプタン] 受容体作動薬との併用治療が臨床的に正当である場合、特に治療開始時および用量増加中は患者を注意深く観察することが推奨されます。デスベンラファクシンとセロトニン前駆体 (トリプトファン サプリメントなど) の併用は推奨されません。
止血を妨げる薬剤(非ステロイド性抗炎症薬、アセチルサリチル酸、ワルファリンなど)
血小板によるセロトニンの放出は止血に重要な役割を果たします。症例対照およびコホート設計に関する疫学研究では、セロトニンの再吸収を妨げる向精神薬の使用と上部消化管出血の発生との関連が実証されています。これらの研究では、非ステロイド性抗炎症薬またはアセチルサリチル酸の併用が出血のリスクを高める可能性があることも実証しました。 SNRI および SSRI をワルファリンと併用すると、出血量の増加などの抗凝固作用の変化が報告されています。ワルファリン療法を受けている患者は、コハク酸デスベンラファキシン療法を開始または中止するときに注意深く監視する必要があります。
エタノール
臨床研究では、デスベンラファクシンがエタノールによって引き起こされる精神的および運動能力の障害を増加させないことが実証されました。ただし、他のすべてのCNS活性薬と同様に、患者にはデスベンラファキシンの服用中はアルコールを摂取しないようにアドバイスする必要があります。
他の薬剤がデスベンラファクシンに影響を与える可能性
CYP3A4阻害剤
CYP3A4 はデスベンラファクシンの除去にはほとんど関与しません。臨床研究では、ケトコナゾール (200 mg を 1 日 2 回) により、濃度対濃度曲線の下の面積が増加しました。デスベンラファクシン (400 mg 単回投与) の時間 (AUC) は約 43% 減少し、相互作用は弱く、 Cmaxは約 8% 減少しました。デスベンラファクシンと強力な CYP3A4 阻害剤を併用すると、デスベンラファクシン濃度が高くなる可能性があります。
他のCYP酵素の阻害剤
in vitroデータに基づくと、CYP 1A1、1A2、2A6、2D6、2C8、2C9、2C19 および 2E1 アイソザイムを阻害する医薬品は、デスベンラファクシンの薬物動態プロファイルに重大な影響を与えるとは予想されません。
デスベンラファクシンが他の薬剤に影響を与える可能性
CYP2D6によって代謝される薬剤
臨床研究では、デスベンラファクシンが 1 日あたり 100 mg の用量で CYP2D6 代謝に臨床的に関連する影響を及ぼさないことが実証されています。コハク酸デスベンラファキシン一水和物を、CYP2D6 基質であるデシプラミン 50 mg の単回投与と組み合わせて、1 日あたり 100 mg の用量で投与すると、デシプラミンの AUC は約 17% 増加しました。 400 mg の用量を投与すると、デシプラミンの AUC は約 90% 増加しました。デスベンラファクシンコハク酸塩一水和物を、モルヒネに代謝されるCYP2D6基質であるコデインの単回60mgと組み合わせて、毎日100mgの用量で投与した場合、モルヒネのAUCは約8%減少した。 CYP2D6 によって代謝される薬物とデスベンラファクシンを併用すると、その薬物の濃度が高くなり、その CYP2D6 代謝物の濃度が低くなる可能性があります。
CYP3A4によって代謝される薬剤
インビトロでは、デスベンラファクシンは CYP3A4 アイソザイムを阻害または誘導しません。臨床研究では、デスベンラファクシン(毎日 400 mg)は、CYP3A4 基質であるミダゾラム(4 mg 単回投与)の AUC を約 31% 減少させました。 2 番目の研究では、コハク酸デスベンラファクシン 50 mg を毎日 4 mg の単回用量のミダゾラムと同時投与しました。ミダゾラムの AUC とCmax は、それぞれ約 29% と 14% 減少しました。デスベンラファクシンと CYP3A4 によって代謝される薬物を併用すると、その薬物への曝露が減少する可能性があります。
CYP2D6 と CYP3A4 の組み合わせによって代謝される薬剤 (タモキシフェンとアリピプラゾール)
臨床研究では、デスベンラファクシン (1 日 100 mg) が、CYP2D6 酵素と CYP3A4 酵素の組み合わせによって代謝される薬物に対して臨床的に関連した効果を及ぼさないことが示されています。

タモキシフェンの単回 40 mg は、主に CYP2D6 によって活性代謝物である 4-ヒドロキシ タモキシフェンとエンドキシフェンに代謝され、CYP3A4 による代謝への寄与はわずかですが、コハク酸デスベンラファキシン一水和物(1 日 100 mg)と組み合わせて投与されました。デスベンラファキシンコハク酸塩一水和物の併用投与により、AUCは3%増加しました。 4-ヒドロキシ タモキシフェンの AUC は 9% 増加しました。エンドキシフェンの AUC は 12% 減少しました。
コハク酸デスベンラファキシン一水和物を、活性代謝物デヒドロアリピプラゾールに代謝されるCYP2D6およびCYP3A4基質であるアリピプラゾールの単回5mgと組み合わせて、1日100mgの用量で投与した。アリピプラゾールの AUC は、コハク酸デスベンラファキシン一水和物の併用投与により 6% 増加しました。デヒドロアリピプラゾールの AUC は、同時投与により 3% 増加しました。
CYP1A2、2A6、2C8、2C9、および2C19によって代謝される薬剤
インビトロでは、デスベンラファクシンは CYP1A2、2A6、2C8、2C9、および 2C19 アイソザイムを阻害せず、これらの CYP アイソザイムによって代謝される薬物の薬物動態に影響を与えるとは予想されません。
P-糖タンパク質トランスポーター
インビトロでは、デスベンラファクシンは P 糖タンパク質トランスポーターの基質でも阻害剤でもありません。
臨床検査との薬物相互作用
デスベンラファクシンを服用している患者において、フェンシクリジン(PCP)およびアンフェタミンの尿免疫学的検査スクリーニング検査で偽陽性が報告されたことが報告されています。これは、スクリーニング検査の特異性が欠如しているためです。デスベンラファクシン療法の中止後、数日間は偽陽性の結果が予想されます。ガスクロマトグラフィー/質量分析などの確認検査により、デスベンラファクシンとPCPおよびアンフェタミンが区別されます。
電気けいれん療法
デスベンラファクシンによる MDD の治療と電気けいれん療法を組み合わせた場合のリスクおよび/または利点を確立する臨床データはありません。
研究室の変更の可能性
デスベンラファクシンコハク酸塩で治療された一部の患者において、血清トランスアミナーゼレベルの軽度の上昇がまれに報告されています(~0.1%および<1%)。その一部はビリルビンレベルの上昇を伴わずに臨床的に有意でした。
脂質
対照研究では、空腹時血清総コレステロール、LDL(低密度リポタンパク質)コレステロール、およびトリグリセリドの上昇が発生しています。これらの異常の一部は、臨床的に重大な可能性があると考えられていました。
所定の閾値を超えた患者の割合を表 1 に示します。
タンパク尿
微量以上のタンパク尿が、固定用量対照研究において観察された(表2を参照)。このタンパク尿は BUN やクレアチニンの増加とは関連しておらず、一般に一過性でした。
電気けいれん療法
デスベンラファクシンによる MDD の治療と電気けいれん療法を組み合わせた場合のリスクおよび/または利点を確立する臨床データはありません。
デベンラファクシンスクシナート一水和物の食物相互作用 – メドレー
これまでのところ報告はありません。
デベンラファキシン一水和物の物質の作用 – メドレー
効果の結果
うつ病の治療法としてのデスベンラファクシンの有効性は、大うつ病性障害の診断基準を満たした成人外来患者を対象とした4件の固定用量プラセボ対照二重盲検無作為化8週間研究と2件の再発予防研究で確立されている。および精神障害の統計 (DSM-IV)。最初の研究では、患者はデスベンラファクシン 100 mg (n = 114)、200 mg (n = 116)、または 400 mg (n = 113) を 1 日 1 回、またはプラセボ (n = 118) の投与を受けました。 2 番目の研究では、患者は 1 日 1 回デスベンラファクシン 200 mg (n = 121) または 400 mg (n = 124) またはプラセボ (n = 124) を投与されました。他の2つの研究では、患者は1日1回デスベンラファクシン50 mg (n = 150およびn = 164)または100 mg (n = 147およびn = 158)またはプラセボ(n = 150およびn = 161)を投与されました。
デスベンラファクシンは、4つの研究で17項目のハミルトンうつ病評価尺度(HAM-D 17 )の合計スコアの改善によって測定され、そのうちの3つの研究では臨床全体印象尺度 – 改善(CGI-I)に基づいて測定されたプラセボよりも優れていることを示しました。 4つの研究。
長期研究では、DSM-IVの大うつ病性障害基準を満たす、デスベンラファクシン50mg/日による8週間の非盲検急性治療に反応し、その後デスベンラファクシンによる12週間安定状態を維持した成人外来患者を、二重のグループに無作為に割り付けた。再発について最大26週間観察し、積極的な治療を続けるかプラセボに切り替えるかを盲目的に選択します。開放相中の反応は、HAM-D 17の合計スコアとして定義されました。 11 で CGI-I ですか? 56日目の評価では2位。安定性は、HAM-D 17でフルスコアを持たないものとして定義されました。オフィス訪問の場合は 16 回。二重盲検段階での再発は次のように定義されました。
- HAM-D 17の合計スコア ?オフィス訪問の場合は 16 回。
- 有効性反応が不十分であるため中止。
- うつ病のため入院。
- 自殺未遂。
- 自殺。
継続的なデスベンラファクシン治療を受けた患者は、プラセボと比較して統計的に有意に再発までの時間が長かった。 26週時点でのカプラン・マイヤー推定再発確率は、デスベンラファクシン治療では14%、プラセボでは30%でした。
2番目の長期研究では、DSM-IVの大うつ病性障害基準を満たし、デスベンラファキシンによる12週間の急性治療に反応した成人外来患者が、急性治療中に投与された患者と同じ用量(200または400mg/日)に無作為に割り付けられた。または再発についてはプラセボを最長 26 週間観察します。開放相中の反応は、HAM-D 17 lt の合計スコアとして定義されました。二重盲検段階での再発は、84 日目の評価で次のように定義されました。
- HAM-D 17の合計スコア ?オフィス訪問の場合は 16 回。
- CGI-I スコア?オフィス訪問の場合は 6 (84 日目と比較)。
- 反応が不十分なため研究を中止。
デスベンラファクシンによる継続治療を受けた患者は、プラセボを受けた患者と比較して、その後の 26 週間の再発率が有意に低かった。
治療結果と年齢および性別との関係を分析したところ、これらの患者の特徴に基づく反応性の差異は示唆されませんでした。これらの研究では、人種が転帰に与える影響を判断するのに十分な情報がありませんでした。
薬理学的特徴
前臨床研究では、デスベンラファクシンが選択的セロトニンおよびノルエピネフリン再取り込み阻害剤 (SNRI) であることが実証されています。デスベンラファクシンの臨床効果は、中枢神経系におけるこれらの神経伝達物質の作用の増加に関連しています。
デスベンラファクシンは、インビトロでムスカリン性コリン作動性受容体、H1-ヒスタミン作動性受容体、またはβ1-アドレナリン作動性受容体を含むいくつかの受容体に対して有意な親和性を持たない。これらの受容体における薬理学的活性は、他の向精神薬で見られるいくつかの抗コリン作用、鎮静作用、および心臓血管作用と関連していることが示唆されています。同じ包括的な結合プロファイリングアッセイにおいて、デスベンラファクシンは、カルシウム、塩化物、カリウム、ナトリウムイオンチャネルを含むいくつかのイオンチャネルに対して有意な親和性を示さなかった。また、モノアミンオキシダーゼ (MAO) に対する阻害活性も示されませんでした。デスベンラファクシンは、インビトロ心臓カリウムチャネル (hERG) 研究では有意な活性を示さなかった。
前臨床げっ歯類モデルにおいて、デスベンラファクシンは、抗うつ作用、抗不安作用、体温調節作用、および疼痛抑制作用の予測活性を実証しました。
薬物動態学的特性
デスベンラファクシンの単回用量の薬物動態は直線的であり、50 ~ 600 mg/日の用量範囲にわたって用量に比例します。平均終末半減期 (t 1/2 ) は約 11 時間です。 1 日 1 回の投与では、約 4 ~ 5 日で定常状態の血漿濃度に達します。定常状態では、デスベンラファクシンの複数回投与の蓄積は直線的であり、単回投与の薬物動態プロファイルから予測可能です。
デスベンラファクシンの薬物動態は、女性と男性で十分に評価されています。性別による違いは最小限でした。全個人のデータを以下に示します。
吸収と分配
デスベンラファキシンコハク酸塩一水和物はよく吸収され、絶対経口バイオアベイラビリティは 80% です。最大血漿濃度までの平均時間(T max )は、経口投与後約7.5時間である。 100 mg の複数回投与後に、それぞれ 6,747 ng.h/mL および 376 ng/mL の AUC およびCmaxが観察されます。
食事の影響
健康な対象者への絶食時および食物存在下(高脂肪食)でのデスベンラファクシンの投与を含む食物存在研究では、食物存在下でCmaxが約 16% 増加する一方、AUC は同等であることが示されました。この違いは臨床的には重要ではありません。したがって、デスベンラファキシンは食事に関係なく摂取できます。
デスベンラファクシンの血漿タンパク質結合は低く (30%)、薬物濃度には依存しません。静脈内投与後の定常状態におけるデスベンラファクシンの分布量は 3.4 L/kg であり、非血管区画への分布を示しています。
代謝と排出
デスベンラファクシンの約 45% は変化せずに尿中に排泄されます。デスベンラファクシンは主に抱合(UGT1A1、UGT1A3、UGT2B4、UGT2B15、UGT2B17 などの UGT アイソフォームによって媒介される)によって代謝され、程度は低いですが酸化代謝によって代謝されます。
投与量の約 19% がグルクロニド代謝産物として排泄されます。尿中の酸化代謝物 (N,O-ジデスメチルベンラファキシン) として 5%。 CYP3A4 は、デスベンラファクシンの酸化代謝 (N-脱メチル化) を媒介する主要なシトクロム P450 アイソザイムです。
高齢者向けの使用
最大300 mgの用量を受けた健康な被験者を対象に実施された研究では、年齢に応じてデスベンラファクシンクリアランスが減少し、300歳以上の被験者ではCmaxが32%増加し、AUC値が55%増加しました。 mg. 18〜45歳の個人と比較して75歳。高齢患者(65歳以上)と若年患者の間で安全性や有効性に関して差は観察されなかったが、一部の高齢患者の感受性が高いことは無視できない。年齢のみに基づいて用量を調整する必要はありませんでした。ただし、使用量を決定する際には、デスベンラファクシンの腎クリアランスの低下の可能性を考慮する必要があります。
小児への使用
18 歳未満の患者に対する安全性と有効性は確立されていません。
腎不全患者
デスベンラファキシンコハク酸塩一水和物 100 mg の薬物動態は、軽度 (n = 9)、中等度 (n = 8)、および重度 (n = 7) の腎不全および透析を必要とする末期腎疾患 (ESRD) を有する被験者 (n = 7) を対象に研究されました。 9)および年齢が一致した健康な対照被験者(n = 8)。クリアランスはクレアチニンクリアランスと有意に相関していた。総体内クリアランスは、健康な被験者と比較して、軽度では29%、中等度では39%、重度では51%、ESRDでは58%減少しました。このクリアランスの減少により、軽度の腎障害のある個人 (24 時間 CrCl = 50 ~ 80 mL/分) では AUC が 42%、中等度の腎障害のある個人 (24 時間 CrCl = 30 ~ 50 mL/分) では 56% 増加しました。108重症者(24 時間の CrCl lt; 30 mL/min)では %、ESRD 患者では 116% でした。
<div cla






-1024x576.jpg?resize=1024,576&ssl=1 "デスロラタジン・ザイダス日興リーフレット")


