

リムパーザ錠の禁忌
オラパリブまたはその配合成分のいずれかに対する過敏症。
リムパーザ錠の薬物相互作用
処方箋なしで入手した薬や漢方薬など、他の薬を服用している場合、または最近使用した場合は、医師または医療専門家に伝えてください。
他の抗がん剤を服用している場合は、この薬を使用しないでください。
一部の薬は、体内のこの薬のレベルに影響を与える可能性があります。この薬は他の薬の作用にも影響を与える可能性があります。以下の物質を含む薬を使用している場合は、医師に知らせる必要があります。
- ケトコナゾール、イトラコナゾール(真菌感染症の治療に使用)
- テリスロマイシン、クラリスロマイシン(細菌感染症の治療に使用)
- リトナビル、ネルフィナビル、インジナビル、サキナビル、ネビラピン(ウイルス感染症、主にHIVの治療に使用)
- リファンピシン、リファペンチン、リファブチン(細菌感染症、主に結核の治療に使用されます)
- フェニトイン、カルバマゼピン、フェノバルビタール(発作およびてんかんの治療に使用)
- モダフィニル(ナルコレプシーと呼ばれる睡眠障害の治療に使用されます)
- セントジョーンズワート(主にうつ病に使用される漢方薬)
これらの薬を使用している場合は、医師または薬剤師に伝えてください。
他の薬を服用している場合は、医師または歯科医に伝えてください。
医師の知識なしに薬を使用しないでください。健康に危険を及ぼす可能性があります。
DNA損傷剤を含む他の抗がん剤とオラパリブを併用した臨床研究では、骨髄毒性の可能性が示されています。 L NPA ZA 単独療法の推奨用量は、他の抗がん剤との併用には適していません。
インビトロのオラパリブは、CYP 1A2、2A6、2B6、2C8、2C9、2C19、2D6、または 2E1 を直接阻害することがほとんどまたはまったくありませんでした。オラパリブを最大 100 uM の濃度で試験した場合には、CYP3A4 の中程度の阻害が観察され、500 uM で試験した場合にはより大きな阻害が見られました。これらの所見は、オラパリブが肝臓および胃腸管において他のCYP3A4基質と臨床的に関連した相互作用を引き起こす可能性があることを示唆しています。オラパリブは、CYP 1A2、2A6、2B6、2C8、2C9、2D6、または 2E1 の時間依存性阻害剤ではありませんでした。これは CYP3A の時間依存性阻害剤であることが示されていますが、重大な臨床効果をもたらす可能性はある程度低いです。 CYP1A2、2B6、および 3A4 の in vitro 誘導が観察され、臨床的に関連するレベルで誘導される可能性が最も高いのは CYP3A4 です。
CYP3A4/5 は、オラパリブの代謝クリアランスに主に関与するアイソザイムです。既知の CYP3A 誘導剤および阻害剤の影響を評価するための臨床研究 (錠剤製剤で実施) では、強力な CYP3A 阻害剤の同時投与により、オラパリブの Cmax が 1.42 倍 (90% CI: 1.33 ~ 1.52) 増加し、平均 AUC が 2.70 ~ 増加することが実証されました。の倍数(90% CI: 2.44-2.97)、強力な誘導剤の同時投与は Cmax を 71% 減少させ(治療指数: 0.29; 90% CI: 0.24-0.33)、平均 AUC は 87% 減少しました(治療指数: 0.13; 90% CI :0.11〜0.16)。したがって、これらのアイソザイムの既知の阻害剤/誘導剤をオラパリブと同時投与しないことが推奨されます。
オラパリブはMDR1の基質ですが、BCRPやMRP2の基質ではありません。 in vitro 研究では、これは MDR1 の阻害剤、BCRP の弱い阻害剤であるが、MRP2 の阻害剤ではないことが示唆されています。オラパリブが MDR1 基質と臨床的に関連した薬物相互作用を引き起こす可能性があります。

オラパリブは、OATP1B1、OCT1、OCT2、OAT3、MATE1、および MATE2K の阻害剤であることも示されています。これらの所見の臨床的関連性は現時点では不明です。オラパリブが OATP1B3 または OAT1 を阻害する可能性は低いです。
オラパリブ食品の効果が研究されました。食物と同時投与すると、オラパリブの吸収速度が低下し(Tmax が 2 時間遅れる)、オラパリブの吸収範囲が増加します(AUC は約 20% 増加します)。
リムパーザ錠の作用
有効性の結果
プラチナ感受性再発卵巣癌
(Ledermann J et al. N Eng J Med 2012;366(15):1382-1392 および Ledermann J et al. Lancet Oncol 2014;15(8):852-861)
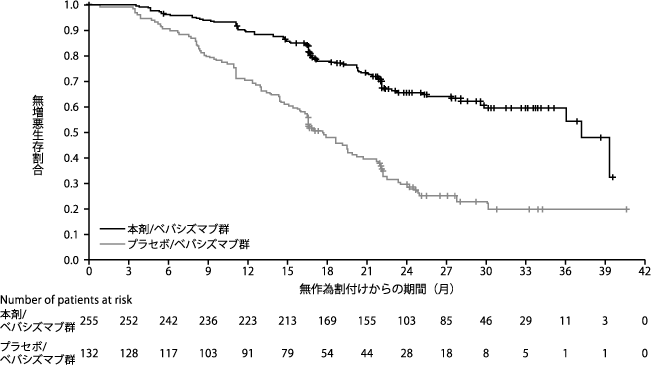
2つ以上のプラチナ含有レジメンによる治療後のプラチナ感受性卵巣癌、原発性腹膜癌、または卵管癌患者の治療における維持療法としてのオラパリブの安全性と有効性が、無作為化第II相二重盲検試験で研究された。 、プラセボ対照(研究19)。この研究では、プラチナベースの化学療法治療終了時に奏効(部分奏効または完全奏効)していた265人のプラチナ感受性患者(オラパリブ136人、プラセボ129人)を対象に、維持療法なしの進行とオラパリブ維持療法の有効性を比較した。奏効は、婦人科がんインターグループ (GCIG) が定義する基準に従って、RECIST および/または CA-125 によって確認されます (最後の治療前のサンプルと比較して CA-125 レベルが少なくとも 50% 減少し、28 日後に確認されます)。患者は、低悪性度(グレード 1)として分類されない漿液性卵巣がん(または漿液性成分)の組織学的診断を受ける必要がありました。主要評価項目は、RECIST 1.0を使用した研究者の評価に基づく無増悪生存期間(PFS)でした。副次評価項目には、全生存期間(OS)、部分/完全奏効+疾患安定として定義される疾患制御率(TCD)、生活の質(HRQoL)、および疾患関連症状が含まれます。最初のその後の治療または死亡までの時間、およびその後の 2 回目の治療または死亡までの時間の探索的分析も実行されました。
プラチナ感受性(最後から2番目のプラチナベースの化学療法終了後6か月以内に疾患が再発)と判定され、化学療法終了後のベースライン(RECISTおよび/またはCA-125による)で部分的または完全な客観的反応を示した患者のみ。最後のプラチナベースの化学療法。患者は、オラパリブまたは他のPARP阻害剤による以前の治療を受けていない可能性があります。ランダム化直前のレジメンを除き、患者は以前にベバシズマブを受けていた可能性がある。オラパリブによる進行後のオラパリブによる反復治療は許可されなかった。
患者は、60週目までは12週間ごとに腫瘍評価を受け、その後はRECISTによって疾患の進行が判定されるか、患者の同意を得て治療が中止されるまで24週間ごとに腫瘍評価を受けた。患者が CA-125 の増加を示した場合(ベースライン CA-125 の 2 倍増加 [ベースラインでの正常値の上限を超えている場合]、または正常値の上限の 2 倍増加 [正常値の上限値を下回っている場合)ベースラインで] 7 日以上の間隔で 2 回)、事前に決められたスケジュール以外で画像評価を受ける可能性がありました。進行が確認されなかった場合、患者は次の腫瘍画像評価まで治療を継続した。したがって、CA-125 測定値の増加に関係なく、患者は RECIST による進行まで治療を維持されました。
この研究は、プラセボと比較した場合、オラパリブ単剤療法維持療法におけるPFSの統計的に有意な改善により主な目的を達成しました(HR 0.35; 95% CI 0.25-0.49; plt;0.00001)。さらに、事前に計画されたサブグループ分析により、BRCA 遺伝子変異を有する卵巣癌患者(N=136、51.3%)が、BRCA 単剤療法による維持療法から最大の臨床効果を示した患者のサブグループであることが特定されました。
BRCA 遺伝子変異のある患者 (n=136) では、無増悪生存期間、最初の治療または死亡までの時間、および 2 回目の治療または死亡までの時間が統計的に有意に増加しました。 PFS中央値はプラセボ中央値より6.9カ月大きかった(HR 0.18; 95% CI 0.10-0.31; plt;0.00001; 中央値11.2対4.3)。ランダム化からその後の最初の治療の開始または死亡までの期間は、オラパリブで治療された患者の方が9.4カ月長かった(HR 0.33; 95% CI 0.22-0.50; plt;0.00001; 中央値15.6カ月対6.2カ月)。ランダム化から2回目の治療開始または死亡までの期間は、オラパリブ治療を受けた患者の方が8.6カ月長かった(HR 0.44; 95% CI 0.29-0.67; p= 0.00013; 中央値23.8カ月対15.2カ月)。全生存期間に統計的に有意な差はありませんでした (HR 0.73; 95% CI 0.45-1.17; p= 0.19; 中央値 34.9 か月 vs 31.9 か月)。
BRCA 遺伝子の生殖系列変異(gBRCA 変異)を持つサブグループ(n=96)では、無増悪生存期間、その後の最初の治療または死亡までの時間、およびその後の 2 回目の治療または死亡までの時間が統計的に有意に増加しました。死。 PFS中央値はプラセボより7.1カ月長かった(HR 0.17; 95% CI 0.09-0.31; plt;0.00001; 中央値11.2対4.1)。ランダム化からその後の最初の治療の開始または死亡までの期間は、オラパリブで治療された患者の方が9.4カ月長かった(HR 0.31; 95% CI 0.19-0.50; plt;0.00001; 中央値15.6カ月対6.2カ月)。ランダム化から2回目の治療開始または死亡までの期間は、オラパリブ治療を受けた患者の方が7カ月長かった(HR 0.43; 95% CI 0.25-0.71; p= 0.001; 中央値22カ月 vs 15カ月)。全生存期間に統計的に有意な差はありませんでした (HR 0.85; 95% CI 0.48-1.52; p= 0.58; 中央値 32.9 か月 vs 30.2 か月)。
BRCA変異およびgBRCA変異の白金感受性卵巣癌患者について認められた研究19の有効性結果の概要を表1に示します。
表 1. BRCA 変異および gBRCA 変異プラチナ感受性卵巣癌患者に対する研究 19 の主な有効性結果の概要
|
a HR= ハザード比 (リスク比)。値 lt;1 はオラパリブに有利です。分析は、治療、以前のプラチナ療法での疾患進行までの時間、以前のプラチナ療法に対する客観的反応、およびユダヤ人の祖先に関する因子を含むコックス比例ハザードモデルを使用して実行されました。 b BRCA変異サブグループのプラセボ治療を受けた患者の約4分の1(14/62; 22.6%)が、その後PARP阻害剤の投与を受けた。 gBRCA変異サブグループのプラセボ治療を受けた患者の約3分の1(13/40; 32.5%)が、その後PARP阻害剤の投与を受けた。 |
BRCA変異集団内では、24週時点での疾患制御率は、オラパリブ群の患者で57%、プラセボ群の患者で24%であった。
患者が報告した症状や生活の質において、治療群間で統計的に有意な差は観察されませんでした。
範囲への影響

オラパリブ錠剤製剤 300 mg を 1 日 2 回複数回投与しても、心臓の再分極に対するオラパリブの臨床的に関連する効果はありません(QT 間隔への効果によって評価)。
薬理学的特徴
薬力学特性
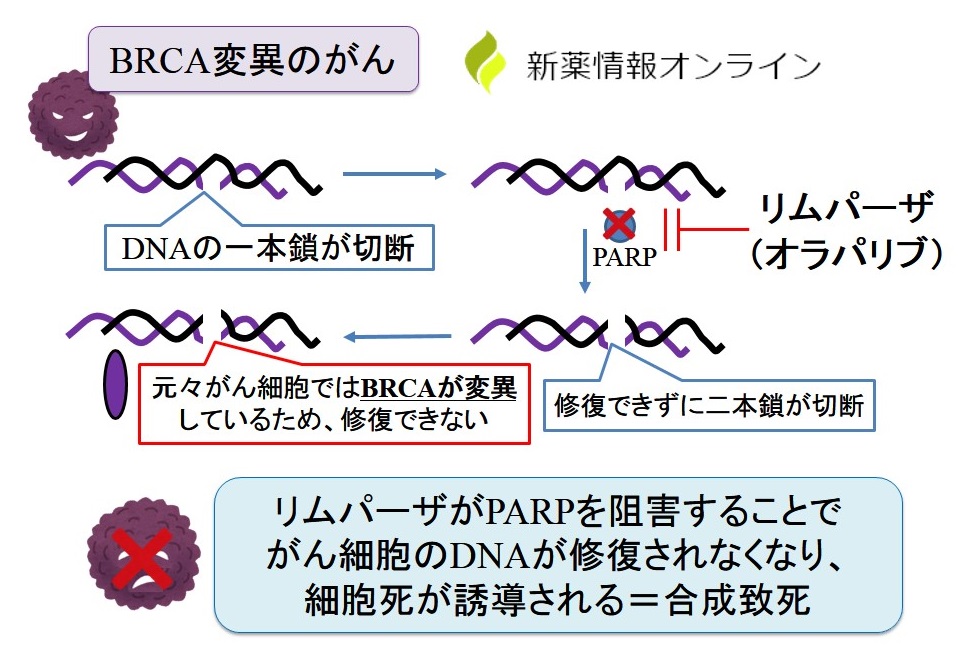
オラパリブは、ヒトポリ(ADP-リボース)ポリメラーゼ(PARP-1、PARP-2、およびPARP-3)酵素の強力な阻害剤であり、選択された腫瘍細胞株のin vitroでの増殖およびin vivoでの腫瘍増殖も阻害することが示されています。単独療法として、または確立された化学療法と組み合わせて投与されます。
PARP は DNA 一本鎖切断を効率的に修復するための重要な酵素であり、PARP 誘発修復の重要な側面の 1 つは、クロマチン修飾後に PARP が自己修飾して DNA から解離して、塩基除去修復酵素へのアクセスを促進する必要があります。オラパリブが DNA に関連する PARP 活性部位に結合すると、PARP の解離を防ぎ、DNA に結合して修復をブロックします。細胞の複製において、複製フォークが PARP-DNA 結合に遭遇すると、これにより二本鎖 DNA の切断が引き起こされます。正常な細胞では、機能的な BRCA1 および 2 遺伝子を必要とする相同組換え修復経路が、この DNA 二本鎖切断の修復に効果的です。しかし、突然変異により機能的な BRCA1 または 2 が欠如している場合、DNA 二本鎖切断は相同組換えによって修復できません。代わりに、非相同末端結合修復経路などの、エラーが発生しやすい代替経路が活性化され、ゲノムの不安定性が増大します。数サイクルの複製の後、ゲノムの不安定性は耐えられないレベルに達し、癌細胞の死をもたらします。これは、がん細胞は正常細胞に比べて DNA 損傷がより多く蓄積しているためです。
BRCA欠損in vivoモデルでは、プラチナ治療後にオラパリブを投与すると、プラチナ治療単独と比較して腫瘍の進行が遅延し、全生存期間が延長した。
薬物動態学的特性
一般的な

1 日 2 回 400 mg の用量でのオラパリブの薬物動態は、約 8.6 L/h の見かけの血漿クリアランス、約 167 L の見かけの分布容積、および 11.9 時間の半減期によって特徴付けられます。
吸収
カプセル製剤中のオラパリブを経口投与した後、吸収は急速であり、一般に投与後 1 ~ 3 時間でピーク血漿濃度に達します。複数回の投与では、関連する蓄積は起こらず、約 3 ~ 4 日以内に曝露の定常状態に達します。
食物との併用投与により、オラパリブの吸収率が低下し(T max が2 時間遅延)、オラパリブの吸収範囲が増加しました(AUC は約 20% 増加)。したがって、患者は食後少なくとも 1 時間以内に L NPA ZA を服用し、投与後 2 時間は食事を避けるべきであることが推奨されます。
分布
オラパリブのin vitroタンパク質結合は、1 日 2 回 400 mg の用量で達成される血漿濃度で約 82% です。
代謝
インビトロでは、CYP3A4 がオラパリブの代謝に関与する主要な酵素であることが実証されました。
女性患者に14C-オラパリブを投与したところ、未変化のオラパリブが血漿中の放射能の大部分(70%)の原因となっており、尿と糞便中に見出される主成分(それぞれ用量の15%と6%)であることが示された。オラパリブの代謝は、主な代謝部位であるカルボキシルシクロプロピル ピペラジン環構造を通じて広範囲に起こりますが、フルオロフェニル環系およびフタラジノン環系を通じてはそれほど広範囲には起こりません。代謝の多くは、その後のグルクロン酸抱合または硫酸抱合を受けて生成される一連の成分との酸化反応に起因すると考えられます。血漿、尿、糞便からそれぞれ約 20、37、20 個の代謝物が検出され、それらのほとんどは測定物質の 1% 以下に相当しました。開いたヒドロキシシクロプロピル環の半分と 2 つの一酸素化代謝産物 (それぞれ約 10%) が主な循環成分であり、一酸素化代謝産物の 1 つが主要な排泄代謝産物でもありました (尿中放射能および糞便放射能のそれぞれ 6% および 5%)。
排除
14C-オラパリブを単回投与した後、投与された放射能の約86%が7日間の収集期間内に回収され、そのうち約44%が尿中に、約42%が糞便中に回収された。物質の大部分は代謝産物の形で排泄されました。
前臨床安全性データ
変異原性
オラパリブは、インビトロでは変異原性の可能性を示さなかったが、哺乳類細胞では染色異常誘発性であった。オラパリブをラットに経口投与すると、骨髄に小核が誘導されました。この染色異常誘発性はオラパリブの既知の薬理と一致しており、ヒトに対して潜在的に遺伝毒性がある可能性があります。
反復投与毒性
ラットおよびイヌにおける最長 6 か月の反復投与毒性試験では、オラパリブの経口投与は良好な忍容性を示しました。両種の主な毒性標的臓器は骨髄であり、末梢血液学的パラメータの変化に関連していた。これらの所見は、臨床的に観察されたものよりも低い曝露で発生し、投与を中止してから 4 週間以内にほとんどが回復しました。ヒト骨髄細胞を使用した生体外研究でも、オラパリブがヒト骨髄細胞に対して細胞毒性があることが確認されました。

生殖毒性学
オラパリブは雄ラットの生殖能力に影響を与えませんでした。メスの生殖能力の研究では、一部の動物で発情の延長が観察されましたが、交尾や生殖能力には影響がありませんでした。この研究では、胚と胎児の生存率が減少しました。
ラットを用いた胚・胎児発育研究において、オラパリブは母体に重大な影響を及ぼさない用量レベルで、胚・胎児生存率を低下させ、胎児体重を減少させ、胎児異常(内臓の変化、骨格異常、重度の眼および脊椎/肋骨奇形を含む)の発生を引き起こした。毒性。
発がん性
オラパリブに関する発がん性研究は行われていません。
QT延長
麻酔をかけた犬を対象とした研究では、最大 15 mg/kg の静脈内投与では QT 延長の証拠はありませんでした。オラパリブは、IC50または226μMでコード化されたhERGカルシウムチャネル試験において活性であり、これは臨床用量での平均ヒト遊離Cmax (1.95μM)より>110倍高い。これは、ヒトにおける QT 延長の可能性が低いことを示唆しています。








